CASE REPORT
https://doi.org/10.47811/bhj.202
First-time use of plasma exchange for atypical hemolytic uremic syndrome in a 5-year-old child in a low-middle-income country: a case report
Jimba Jatsho1, Sonam1, Sonam Choden2
1Department of Pediatrics, Gyaltsuen Jetsun Pema Wangchuck Mother and Child Hospital
2Samtse General Hospital
Corresponding author:
Dr Jimba Jatsho
jimbajatsho@gmail.com
ABSTRACT
Atypical hemolytic uremic syndrome (aHUS) is a rare, life-threatening thrombotic microangiopathy characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury, caused by dysregulation of the alternative complement pathway. We report the first documented case of aHUS managed with plasma exchange (PLEX) in Bhutan. A previously healthy 5-year-old girl presented with fever, hematuria, severe anemia, thrombocytopenia, and renal dysfunction. Secondary causes were excluded and she was diagnosed with aHUS. Due to unavailability of complement inhibitors, she was managed with corticosteroids, peritoneal dialysis, transfusions, and delayed initiation of PLEX. Following PLEX, hematologic parameters and urine output improved, allowing dialysis discontinuation after 24 days. Renal biopsy revealed focal cortical necrosis with partial renal recovery. She remained in intensive care for 27 days, followed by transfer to the pediatric ward and discharge four days later. At two-month follow-up, she remained dialysis-independent with stable but impaired renal function, highlighting management challenges in resource-limited settings.
Keywords: Acute kidney injury; atypical hemolytic anemia syndrome; hemolytic anemi; thrombocytopenia; thrombotic microangiopathiesia
INTRODUCTION
Atypical hemolytic uremic syndrome (aHUS) is a rare form of thrombotic microangiopathy (TMA) caused by dysregulation of the alternative pathway (AP) of the complement system. It is characterized by a triad of thrombocytopenia, acute kidney injury (AKI), and microangiopathic hemolytic anemia (MAHA) (1). The global epidemiology of aHUS is not well known. However, some studies report a prevalence of 3.3 per million in children under 18 years of age (2). aHUS carries a poor prognosis, with mortality reported in approximately 25% of cases and progression to end-stage kidney disease (ESKD) occurring in as many as 50% of affected individuals (3). In high-resource settings, treatment options include eculizumab, a monoclonal antibody that inhibits terminal complement activation. Although the use of eculizumab has revolutionized outcomes, disparities in global access remain a major barrier. Therefore, plasma exchange (PLEX) and immunosuppression remain the primary modality of treatment in developing countries. Here, we present the first documented case of aHUS managed with PLEX in Bhutan, emphasizing the challenges and outcomes in a resource-limited setting.
PATIENT INFORMATION
A 5-year-old girl with no significant past medical history presented with fever, vomiting and cough for 3 days associated with hematuria for 2 days to the Central Regional Referral Hospital in Gelephu. There was no history of hematemesis, hemoptysis or neurological symptoms. She also had no prior history of bloody diarrhea.
CLINICAL FINDINGS
On general examination, the child looked ill, with altered sensorium and severe pallor. Systemic examination revealed bilateral crepitations with reduced air entry on the right side, along with abdominal distension.
DIAGNOSTIC ASSESSMENT
She was found to be severely anaemic (haemoglobin 5.3 g/dL), thrombocytopenic (platelets 15,000/μL), and had acute kidney injury (creatinine 2.8 mg/dL). Her peripheral smear revealed schistocytes suggestive of microangiopathic hemolysis, with elevated lactate dehydrogenase (LDH) 2345 U/L, ferritin >2000 ng/ml, and a negative Coombs test. Serum Complement C3 was low (44.7 mg/dl) with hypoalbuminemia (serum albumin 2.51g/dl).
Tropical infections like malaria, scrub and dengue, were ruled out. Viral markers were negative. Urinalysis showed proteinuria and microscopic haematuria. Blood, urine, pleural fluid, and stool cultures were all negative.
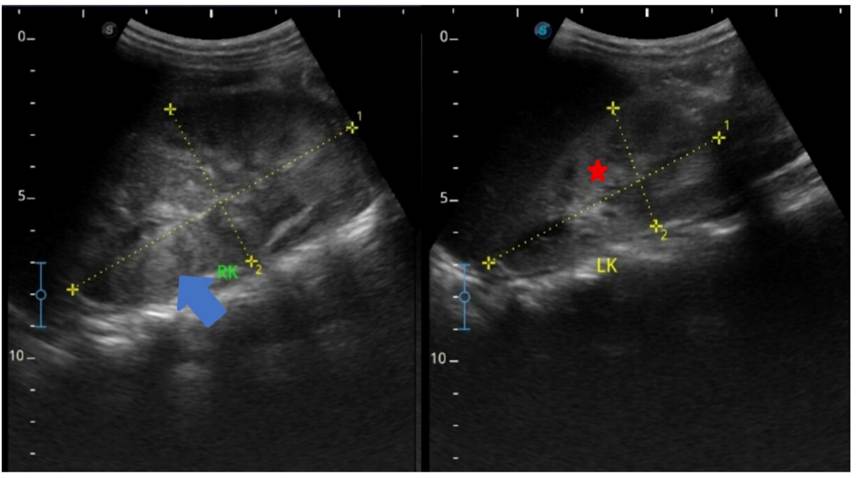
Figure 1. Ultrasound of the kidneys showing enlarged right kidney (10 x 5.6cm) with bilateral increased renal cortical echoes (blue arrow) with poorly maintained cortico-medullary differentiation (red star).
ADAMTS13 (A Disintegrin and Metalloproteinase with a Thrombospondin Type 1 motif, member 13) activity and anti- complement factor H (CFH) antibody testing was not available locally and were not performed.
Point-of-care ultrasound (POCUS) of the chest and kidneys revealed significant bilateral B-lines; a positive shred sign in the posterior right lower zone with a 0.4 cm pleural effusion; and right renomegaly with bilateral increased renal echogenicity and loss of corticomedullary differentiation
Genetic testing using next generation sequencing for hereditary HUS panel was done and noted to be negative. A targeted next-generation sequencing panel for hereditary HUS was performed, including genes implicated in complement-mediated TMA: CFH, CFI, CD46 (MCP), C3, CFB, THBD, DGKE, and the CFHR1-CFHR5 gene cluster.
Renal biopsy revealed focal cortical necrosis affecting 8-10% of the kidney with evidence of regenerative activity. The viable glomeruli demonstrated ischemic changes with approximately 29.4% of sampled glomeruli demonstrating segmental tuft sclerosis. Additional findings included patchy acute tubular injury, mild chronic interstitial inflammation with striped tubulointerstitial fibrosis, and hemosiderin deposits in the interstitium, consistent with thrombotic microangiopathy.
THERAPEUTIC INTERVENTION
Initially she was managed with antibiotics for her pneumonia but in view of rapid clinical and renal deterioration, methylprednisolone pulses were administered for 3 days. She received 3 units of packed red cells and 2 units of platelet. Acute peritoneal dialysis (PD) was subsequently started on the 3rd day of admission due to oliguric acute kidney injury. PLEX could only be started on day 7 of admission. Daily PLEX of 35 ml/kg per session was initiated for one week, followed by alternate-day sessions for next two sessions for a total of 9 cycles (Figure 2). A combination of albumin and fresh frozen plasma (FFP) was used as replacement for the PLEX. No complications were noted during this procedure.
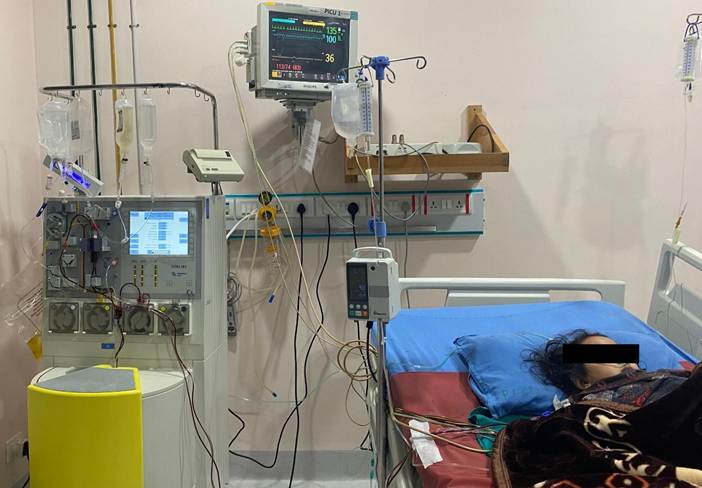
Figure 2. Therapeutic plasma exchange (PLEX) being performed in the pediatric intensive care unit using a Fresenius COM.TEC apheresis machine.
FOLLOW - UP AND OUTCOMES
Following initiation of PLEX, her urine output improved and haematological parameters for haemolysis showed an improving trend, hence PLEX was tapered and stopped after 9 cycles. Supportive care with antihypertensives, volume management, and nutritional support was continued. PD was continued for 24 days and subsequently discontinued following recovery of urine output and overall laboratory and clinical improvement
On the last follow up, 8 months post-onset, her creatinine was 2.14 mg/dl, Urea 98 mg/dl, serum albumin 4.2 g/dl, LDH 402 U/l, Hb 11.8 g/dl, platelets 274000/μL, and ferritin 884.4 ng/ml. She remains non-oliguric and her blood pressure is controlled <90th centile.
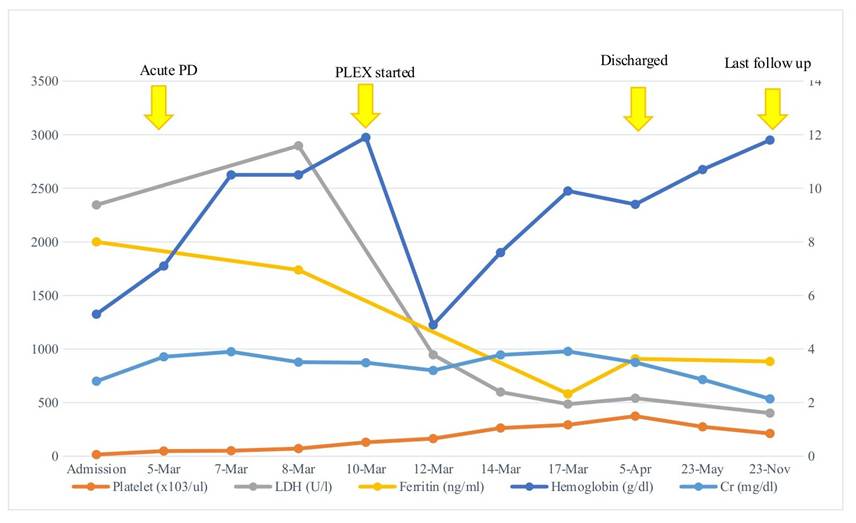
Figure 3. Line graph representing trend of key laboratory parameters from admission to follow up.
DISCUSSION
Atypical hemolytic uremic syndrome (aHUS) is a rare and severe form of thrombotic microangiopathy (TMA) that predominantly affects the kidneys, characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury (AKI) (3). Unlike typical HUS, which is commonly associated with Shiga toxin-producing Escherichia coli infections, aHUS result from uncontrolled activation of the alternative complement pathway, often due to genetic mutations or acquired autoantibodies against complement regulatory proteins (4,5) Early diagnosis and targeted treatment are essential to prevent progression to end-stage kidney disease and to reduce mortality.
The diagnosis of aHUS is particularly challenging in resource-limited settings. It requires the exclusion of other TMAs such as Shiga toxin-associated HUS, thrombotic thrombocytopenic purpura (TTP), and secondary TMAs linked to infections, autoimmune diseases, malignancies, or medications (6). Specific investigations, including ADAMTS13 activity testing, complement factor assays, and genetic analysis, are central to confirming the diagnosis and guiding therapy (7). However, in low- and middle-income countries (LMICs), including Bhutan, these tests are unavailable locally and must be sent abroad, leading to significant delays and increased costs. As seen in our case, clinicians often have to rely on clinical acumen and exclusion of more common aetiologies to make a presumptive diagnosis of aHUS.
Management of aHUS has been revolutionized by the introduction of terminal complement inhibitors, particularly eculizumab and ravulizumab, which have shown dramatic improvements in renal outcomes and survival (8). These are now considered the standard of care in high-income countries; however, their high-cost limits accessibility in most LMICs. Large case series from India have shown improved outcomes with prompt plasma exchange and immunosuppression in anti-factor H autoantibody-associated aHUS, supporting plasma therapy as a pragmatic option where complement inhibitors are unavailable (9). In Bhutan and similar settings, plasma exchange or plasma infusion remains the mainstay of treatment, despite being less effective and associated with higher risks of complications. Furthermore, plasma therapy requires substantial infrastructure including the availability of fresh frozen plasma, expertise in apheresis, and critical care monitoring that may not always be consistently accessible (10).
Our case illustrates the crucial challenges in the diagnosis and management of aHUS. First, the lack of timely access to diagnostic testing delays confirmation of aHUS and initiation of appropriate therapy. Second, even when plasma therapy is initiated, logistical constraints in maintaining adequate supply and expertise can compromise care. Third, the unavailability of complement inhibitors from therapeutic options highlights the disparity in access to life-saving treatments for rare diseases in LMICs. Additionally, without complement factor studies or autoantibody testing, the exact pathophysiology (genetic mutation vs. autoantibody-mediated) remains unknown, which has implications for recurrence risk and family counselling.
To address these gaps, several pragmatic strategies could be implemented. Strengthening the management of complement-mediated thrombotic microangiopathy in Bhutan requires structured regional collaboration and targeted capacity building. Establishing telemedicine-based case discussions with regional pediatric nephrology centers could enable early diagnostic support and treatment guidance, particularly for complex TMA cases. Engagement with international rare disease and complement-focused global health initiatives may facilitate access to complement inhibitors through compassionate-use or tiered pricing programs. Furthermore, strengthening pediatric nephrology services and capacity-building initiatives to enhance expertise in the diagnosis and management of rare kidney diseases are essential steps toward improving outcomes (11).
In parallel, Bhutan could develop stepwise local diagnostic capacity by standardizing TMA diagnositic protocols, prioritizing ADAMTS13 activity and anti-factor H antibody assays testing, followed by phased introduction of targeted next-generation sequencing panels through regional laboratory partnerships. Given Bhutan's small population, a centralized national TMA diagnostic and treatment pathway is feasible and could serve as a model for rare disease care in similar low-resource settings.
In conclusion, this case highlights the complexities of managing aHUS in resource-limited settings such as Bhutan. Despite delayed initiation of plasma exchange, the child ultimately achieved dialysis independence. In a resource-limited setting where long-term access to chronic dialysis is constrained, this outcome represents not only clinical recovery but also a profound impact on the child's long-term quality of life and family burden. Addressing diagnostic and therapeutic challenges through collaborative, innovative, and sustainable approaches is critical to ensuring equitable care for children with this rare but devastating condition.
INFORMED CONSENT
Written informed consent for publication of de-identified clinical details and images was obtained from the patient's parents.
ACKNOWLEDGEMENT
We would like to thank the patient's parents for allowing us to use de-identified clinical details and images for publication purpose.
REFERENCES
1. Yan K, Desai K, Gullapalli L, Druyts E, Balijepalli C. Epidemiology of atypical hemolytic uremic syndrome: a systematic literature review. Clin Epidemiol. 2020;12:295-305. [PubMed] [Full Text] [DOI]
2. Zimmerhackl LB, Besbas N, Jungraithmayr T, et al. Epidemiology, clinical presentation, and pathophysiology of atypical and recurrent hemolytic uremic syndrome. Semin Thromb Hemost. 2006;32(2):113-20. [PubMed] [DOI]
3. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676-87. [PubMed] [Full Text] [DOI]
4. Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60. [PubMed] [Full Text] [DOI]
5. Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2014;25(1):55-64. [PubMed] [Full Text] [DOI]
6. George JN, Nester CM. Syndromes of Thrombotic Microangiopathy. N Engl J Med. 2014;371(7):654-66. [PubMed] [Full Text] [DOI]
7. Fakhouri F, Zuber J, Fremeaux-Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet. 2017;390(10095):681-96.[PubMed] [DOI]
8. de Souza RM, Correa BHM, Melo PHM, Pousa PA, et al. The treatment of atypical hemolytic uremic syndrome with eculizumab in pediatric patients: a systematic review. Pediatr Nephrol. 2023;38(1):61-75. [PubMed] [Full Text] [DOI]
9. Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014;85(5):1151-60. [PubMed] [Full Text] [DOI]
10. Hussein G, Liu B, Yadav SK, Warsame M, Jamil R, Surani SR, Khan SA. Plasmapheresis in the ICU. Medicina (Kaunas). 2023;59(12):2152.[PubMed] [Full Text] [DOI]
11. Kamath N, Erickson RL, Hingorani S, Bresolin N, Duzova A, Lungu A, Bjornstad EC, Prasetyo R, Antwi S, Safouh H, Montini G, Bonilla-Felix M. Structures, Organization, and Delivery of Kidney Care to Children Living in Low-Resource Settings. Kidney Int Rep. 2024;9(7):2084-95. [PubMed] [Full Text] [DOI]
|
AUTHORS CONTRIBUTION Following authors have made substantial contributions to the manuscript as under: JJ: Conceptualization, writing original draft, reviewing, editing, data collection, data analysis, method development, funding acquisition S: Conceptualization, method development, reviewing, editing SC: Manuscript reviewing, editing Authors agree to be accountable for all respects of the work in ensuring that questions related to the accuracy and integrity of any part of the work are appropriately investigated and resolved. |
|
CONFLICT OF INTEREST |
|
None |
|
GRANT SUPPORT AND FINANCIAL DISCLOSURE |
|
None |